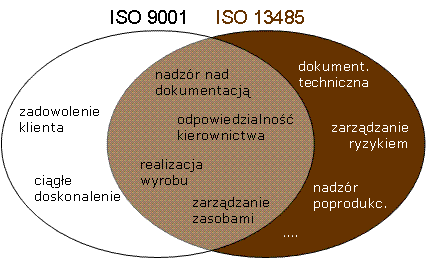
Producenci wszystkich klas wyrobów medycznych mają obowiązek wdrożyć i stosować system zarządzania jakością. Dodatkowo, system jakości jest auditowany przez jednostkę notyfikowaną na potrzeby certyfikacji wyrobów wyższych klas (ocena według załącznika IX, X(A) rozporządzenia MDR). Dla systemów jakości w sektorze wyrobów medycznych normą zharmonizowaną jest EN ISO 13485 (w Polsce dostępna jako PN-EN ISO 13485). Poza wymaganiami typowymi dla systemów zarządzania jakością (znanymi np z ISO 9001), norma ta reguluje wiele kwestii formalnych, określonych w Rozporządzeniu MDR, takich jak:
- dokumentacja techniczna
- odniesienie do wymagań prawnych
- zarządzanie ryzykiem wyrobu
- szczegółowe wymagania dotyczące realizacji wyrobu, środowiska i uprawnień personelu
- szczegółowe wymagania dotyczące wyrobów o ograniczonym terminie użycia lub specjalnych warunków przechowywania
- system informacji zwrotnej
- raportowanie incydentów
- przechowywanie dokumentów przez czas życia wyrobu
Przygotowanie do certyfikacji
System ISO 13485 wprowadzają najczęściej producenci wyrobów medycznych na potrzeby oceny zgodności z MDR, ale mogą go również stosować inne firmy z branży wyrobów medycznych, np firmy serwisowe czy dostawcy krytycznych komponentów dla producentów sprzętu medycznego. Co ciekawe, obecnie wymagane jest także posiadanie certyfikowanego systemu m.in. przez dystrybutorów, którzy zajmują się tłumaczeniem instrukcji wyrobów.
Wdrażanie systemu zarządzania jakością według ISO 13485 odbywa się w podobny sposób, jak innych systemów jakości, np systemów wg. popularnej normy ISO 9001. Zwykle wstępnym etapem wdrożenia jest ocena aktualnych rozwiązań przyjętych w firmie – audit wstępny. Ma on na celu określenie, jakie wymagania normy muszą być uzupełnione. W firmie, w której dotychczas nie funkcjonował system jakości, identyfikuje się procesy główne i pomocnicze, określa się ich współzależności i podstawowe metody nadzorowania tych procesów. Równolegle tworzy się niezbędną dokumentację: począwszy od polityki i celów jakości, poprzez księgę jakości i procedury, aż po instrukcje robocze. Szczegółowość dokumentacji zależy od produkowanych wyrobów, procesów realizowanych przez firmę i innych czynników, takich jak wielkość firmy czy kompetencje pracowników.
W odróżnieniu od ISO 9001 norma ISO 13485 jest znacznie mniej elastyczna pod względem minimalnej dokumentacji. Bardzo wiele obszarów, zwłaszcza tych specyficznych dla przemysłu medycznego musi być określonych w udokumentowanych procedurach. Dotyczy to takich zagadnień jak:
- Nadzór nad dokumentami i zapisami
- Projektowanie i rozwój
- Zakupy
- Nadzorowanie produkcji
- Monitorowanie i pomiary
- Monitorowanie i nadzorowanie środowiska pracy
- Identyfikacja i identyfikowalność
- Serwis
- Zabezpieczanie wyrobu
- Nadzór nad wyrobem niezgodnym / zwróconym
- Nadzorowanie wyrobu o ograniczonym czasie lub wymagającego specj. warunków
- Walidacja oprogramowania
- Walidacja sterylizacji
- System informacji zwrotnych
- Notatki doradcze
- Raportowanie incydentów
- Audity wewnętrzne
- Zbieranie i analizowanie danych
- Działania korygujące i zapobiegawcze
Opracowanie dokumentacji to tylko część wdrożenia systemu – celem jest zapewnienie, że przyjęte ustalenia będą realizowane w praktyce. Aby sprawdzić, czy taka zgodność jest zapewniona, przeprowadza się audity wewnętrzne. Ważnymi źródłami informacji są zapisy – wszelkiego rodzaju dokumenty tworzone w ramach bieżącej działalności, np raporty serii, raporty badań itp.
Wdrożenie systemu można uznać za zakończone po wykonaniu działań korygujących z niezgodności ujawnionych podczas auditów.
W przypadku obsługi przez CEMED.INFO sp. z o.o. przygotowanie do certyfikacji uznajemy za zakończone dopiero po udzieleniu firmie certyfikatu przez jednostkę notyfikowaną. Firma nasza oferuje kompleksową obsługę we wdrażaniu systemów ISO 13485, w tym zaplanowanie, udokumentowanie, weryfikację systemu jakości. Istotną częścią wdrożenia są szkolenia dla pracowników.
[Sprawdź – kompleksowa obsługa wytwórców wyrobów medycznych]